

Myka is a neuroscientist and immunologist whose research focuses on risk factors and interventions for neurodevelopmental disorders. Her work has been published in Science, Nature Reviews Neuroscience, and Immunity. She currently manages a research lab focused on children with profound neurodevelopmental disorders and publishes the Journal Club with Myka Substack. She’s also in the process of launching an independent bookstore.
Spring is finally coming to Canada.
It never shows up all at once, just a flicker at first. A slant of sunlight across the kitchen table, river water rustling beneath the winter ice. It’s like this every year. And yet the long winter always makes me doubt spring will come again.
In Greek myth, Demeter is the goddess of the harvest. When her daughter, Persephone, is taken to the underworld by Hades, Demeter’s grief halts the growth of all things. A deal is eventually struck: Because she ate a few pomegranate seeds in the underworld, Persephone must spend part of each year below but will be allowed to return to Demeter every spring. Even knowing this, Demeter mourns her absence as if the return were never guaranteed.
By the end of winter, I am Demeter in abeyance, disbelieving the cycle. As if the cold will stay forever; as if the underworld has won.
But today, I stand corrected. One warm day is all it takes. The soil softens. Possibility stirs.
Science can feel like that, too.
We work inside inherited frameworks, mistaking the scaffolding of our understanding for reality itself. And then, sometimes, a shift. A sliver of evidence cracks through the surface and what once felt certain begins to thaw.
In neuroscience and immunology, just such a crack appeared in 2007. Suddenly, some conditions doctors had long diagnosed as psychosis, personality changes, cognitive decline, or just psychiatric illness, appeared instead to be effects of the immune system attacking the brain.
What had scientists discovered? A specific kind of autoimmune attack. Patients were making antibodies that targeted the NMDA receptor—a critical junction for passing information between neurons (synaptic communication). The disorder was eventually dubbed anti-NMDA receptor encephalitis, and later, when other antibodies with other targets were discovered able to cause similar conditions, autoimmune encephalitis.
The discovery marked more than a new diagnosis: namely, it challenged a foundational belief shared by both neuroscience and immunology. For most of modern science, the brain was considered “immune privileged”—a kind of no-fly zone for immune cells. The idea was that these two systems, though essential, operated on opposite sides of a biological firewall (the blood-brain barrier).
But that firewall turns out more porous than we thought. In the past several decades, neuroscience and immunology have come to a growing recognition that the immune system doesn’t just defend the body from infections. It also shapes, regulates, and sometimes disrupts what we think of as mental life.
The Pre-History of Autoimmune Encephalitis
The identification of autoimmune encephalitis was the culmination of more than 40 years of mysterious clinical observations: patients with seizures, memory loss, and mood changes who were later found to have cancer. The illness first showed up in the medical literature in 1960, when clinicians described three adults with what they called “subacute encephalitis of later adult life mainly affecting the limbic areas.”
If we’re being honest, the whole diagnosis is basically medical hand-waving. “Subacute” just means “not super-fast, but not slow either,” “encephalitis” means “something’s inflamed in the brain,” and “limbic areas” refers to a fuzzy collection of brain bits involved in emotion and memory. In other words, something weird is happening in the emotional memory parts of the brain, kind of suddenly, and we have no idea why. Not exactly diagnostic precision, but it was a start.
Many patients with this condition deteriorated rapidly, and a significant number were only identified post-mortem, when pathologists would find limbic inflammation, neuronal loss, and immune cell infiltrates—often alongside “occult” (literally, hidden) tumours like small-cell lung cancer or testicular germ-cell tumours.
Eventually, this cluster of symptoms came to be known as paraneoplastic limbic encephalitis, a term that, again, sounds more precise than it is. It basically meant: there’s inflammation in the emotional-memory circuits of the brain, and oh look, the patient also had cancer.
The prevailing theory at the time was that certain tumours were triggering an immune response that accidentally spilled over into the nervous system. But the antibodies identified in these patients weren’t targeting the outside of cells, where they could interfere with function. Instead, they recognized intracellular proteins, buried deep inside neurons. And antibodies, as any immunologist will tell you, can’t easily get inside living cells.
That made the antibodies unlikely culprits. Before 2007, they were thought to be a consequence of the disease—evidence that the immune system had already done damage. According to this model, it was a broader inflammatory process, most likely involving cytotoxic T cells, that was attacking neurons first. When those neurons died, they spilled their internal contents into the surrounding tissue, including proteins the immune system had never seen before. Then antibodies were generated against those proteins.
In other words, the antibodies were downstream, not upstream. These antibodies weren’t early warning signs or causes; they were immunological debris of damage done.
The figure below illustrates this hypothesized model: a tumour triggers a general immune activation, leading to neuronal cell death, and only then do antibodies against intracellular proteins appear—not as instigators, but as clues left at the scene.
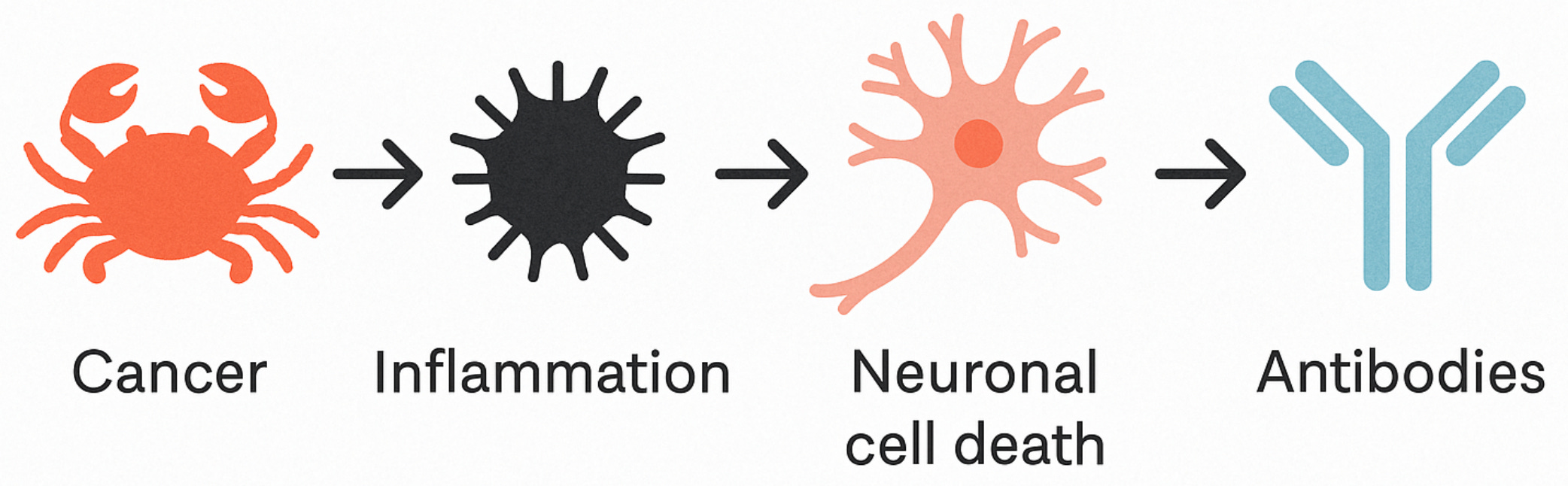
Treatment efforts were mostly ineffective. By the time doctors figured out what was going on, neurons were often already lost. And because symptoms frequently came before a cancer diagnosis, the connection wasn’t always obvious. Patients were often misdiagnosed with psychiatric illness, admitted to the wrong ward, and left to decline. You can imagine how many cases were missed.
So, for decades, neurologists were stuck with fragments: a mysterious syndrome that attacked personality and memory, an occasional tumour, a few tantalizing antibodies, and a vague sense that the immune system was somehow involved. But nothing that tied it all together.
Then, in 2007, a change in scientific technique changed the narrative.
Previously, researchers used fixed tissue slices or biochemical assays which were enriched for proteins inside of cells (intracellular proteins) to study the disease. But in 2007, researchers using rat brain sections and live cultured neurons—techniques that preserved the delicate structure of the cell surface—observed antibodies binding in real time to the outside of neurons, where they could actually interfere with synaptic function. So, these newly identified antibodies weren’t targeting proteins buried inside cells. They were on the surface, targeting a receptor that’s essential for how neurons communicate: the NMDA receptor, a key player in learning and memory.
And that changed everything. If you remember one of the basic rules for identifying causes in science—what epidemiologists call temporality—then you’ll see why this matters: a true cause has to come before the effect. An antibody that alters the properties of a neuron’s signalling channels is very likely to be the cause, not the consequence, of cognitive impairment.
These antibodies weren’t just markers anymore: they were agents of causality, the kind of smoking gun every translational scientist dreams of finding. They didn’t kill neurons outright; they disrupted synaptic function, impairing signalling.
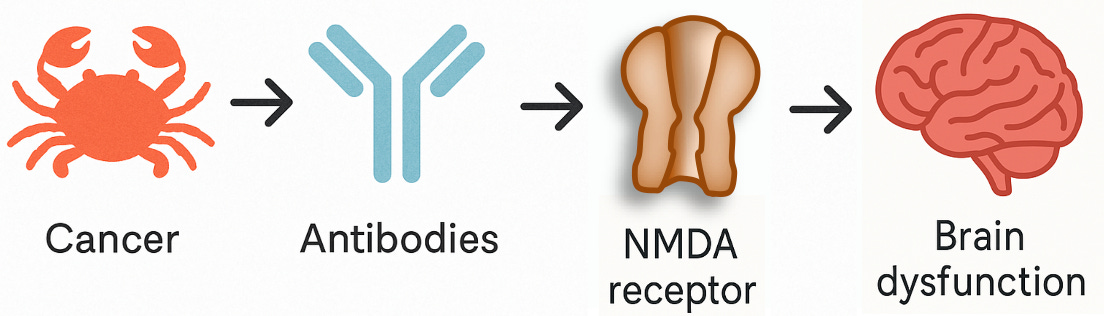
For the first time, medicine had uncovered a psychiatric syndrome caused by a treatable autoimmune process. And here’s the extraordinary part: the damage was reversible. When doctors treated the immune response and, where applicable, removed the tumour, patients came back to their former selves.
Once the basics of autoimmune encephalitis were understood, interesting variations were identified. Many new cases—especially in children and young adults—had no tumours at all. The initial cancer link had cracked open the mystery, but the real story was broader: an autoimmune attack, often in people with personal or family histories of autoimmunity, sometimes following infection, that targeted the brain and produced dramatic changes in behaviour and cognition.
The discovery of autoimmune encephalitis landed like an epistemic shockwave, forcing the field to reckon with just how much it had overlooked.
But it was also something else, something rare and worth celebrating: a clear win for translational neuroscience. A field that’s more Mets than Yankees when it comes to its win–loss record—plenty of grit, a lot of heartbreak, and far more swings and misses than walk-off victories. But here, finally, was an extraordinary win. A baffling clinical syndrome with a patient population hiding in plain sight. Eventually, a molecular mechanism uncovered and a treatment strategy that worked. And best of all, patients who, if diagnosed early enough, recovered.
If you followed my previous series on Alzheimer’s disease, you’ll know how rare this kind of clarity is in neurology.
You’d like to think these revolutions in medical comprehension would spread instantly to specialists worldwide, but that rarely happens. In the case of autoimmune encephalitis, it took more than recognition and publication in academic papers—it took a compelling story. In 2012, journalist Susannah Cahalan published Brain on Fire, a first-person account of her descent into apparent psychosis that turned out to be autoimmune encephalitis. Her memoir exposed a hidden epidemic; at the time, only about 10 percent of cases were being correctly diagnosed. But it also exposed something else: the power of how we think about biological systems to inhibit our ability to understand them.
The Double-Edged Sword of Metaphor
For most of the 20th century, we described the immune system as a military force, defending the borders of the body against invasion. The brain was considered “immune privileged”—too precious for warfare, sealed off by evolution from immune influence. This metaphor wasn’t just poetic; it shaped what we looked for, what we measured, and what we could see.
But reality—as ever—resists neat conceptual packaging.
We now know the immune system isn’t just a warrior—it’s a curator, a gardener, a maintainer of order, and a mediator of meaning throughout the body, including the brain.
In this series, we’ll explore how the discovery of autoimmune encephalitis has pushed us to rethink not just the relationship between the brain and immune system, but how biological systems interact, how diseases emerge, and how our frameworks for understanding can both illuminate and obscure the truth. Throughout these discussions, we’ll return to the role of language and metaphor in science—how the words and models we choose shape what questions we ask, what experiments we design, how we interpret those results and ultimately what we can discover.
The military metaphors that dominated immunology for a century weren’t wrong, exactly. They were incomplete. They were useful except when they weren’t. They helped us understand one aspect of immune function while obscuring others. This pattern—of metaphors both revealing and concealing—runs through the history of science. We need models, categories, poetic association and even myths to make sense of complexity, but we also need the flexibility to step outside them, to remember that naming is not the same as knowing.
500 Million Years of Your Friendly and Fearsome Immune System
By the time you finish reading this sentence, your immune system will have surveilled, evaluated, and responded to millions of signals within your body. Even when you’re sitting listlessly on the sofa, mad at yourself for doing “nothing,” your immune system is busy monitoring trillions of microbes living in your gut, responding to countless particles in the air you breathe, and maintaining constant surveillance of every tissue in your body. This is both amazing and a matter of survival.
But the very sophistication that keeps us alive can turn against us, and for a growing number of people, this remarkable system becomes a source of danger. Autoimmune disorders now affect nearly 17 million people in North America, or roughly 1-in-20. Different autoimmune conditions tend to emerge at different times in life: Type 1 diabetes in childhood, lupus and multiple sclerosis in early adulthood, and rheumatoid arthritis in middle age. And now we’re discovering that conditions once thought entirely psychiatric or neurological—certain cases of psychosis, memory loss, personality changes, seizures, and dementia—can be caused by the immune system attacking the brain.
What other conditions might we still fail to recognize as immune-related? Are autoimmune disorders in fact separate diseases, or do they cluster in different manifestations of broader patterns we’re only beginning to recognize? These questions are fundamental to a better understanding of why autoimmunity occurs, and more importantly, how to prevent and treat it. To get closer to that understanding, we need to look at how our immune system evolved.
Take It Back to When Life Got Complicated
The story begins with an ancient accident that gave vertebrates their most powerful defense system. It’s a story that helps explain both why our immune system is remarkably effective and why it sometimes turns against us. More crucially, it’s a story that matters to anyone who might develop an autoimmune condition—which, given current trends, could be any of us (except me, because I already have a few).
Somewhere in the ancient oceans of the Cambrian era, before dinosaurs, before mammals, when trilobites were finding their legs and vertebrates were developing backbones, something extraordinary happened inside a primitive vertebrate. A piece of rogue DNA, a transposable element (think: molecular hitchhiker with a crowbar), inserted itself into the genome near genes involved in recognizing pathogens.
Transposable elements (transposons) are selfish bits of DNA—genomic drifters that copy and paste themselves across genomes and species. (Richard Dawkins popularized the idea of “selfish genes” in the 1970s; Francis Crick and Leslie Orgel later extended it to DNA itself.) These sequences don’t prioritize serving the organism that hosts them. They serve themselves, hitching rides through evolutionary time by being exceptionally good at getting duplicated.
Where do they come from? Most likely from viruses or ancient bacteria, early genomic parasites that learned how to hack into host DNA and stay there. We don’t know exactly who left the calling card that became our essential immune gene; we only know what they left behind: a pair of enzymes capable of cutting and rearranging DNA with extraordinary precision.
What of consequence do they do? Usually, nothing. They insert, and that’s it. They’re eventually silenced, deleted, or eroded by time. But this one stuck and it rewired the immune system. This was the accident that would make complex vertebrate life possible.
This single insertion became the cornerstone of adaptive immunity—the sophisticated defense system that allows vertebrates to survive childhood, heal from injuries, fight off infections, and live long enough to develop complex social bonds and sophisticated brains. Without it, there would be no long-lived vertebrates, no social mammals, no human culture. A chance genetic event became the gateway to biological complexity.
That bit of DNA became what we now call RAG—short for Recombination Activating Gene. What it activated was nothing short of the immunological big bang.
Why the Cambrian? Multicellular life was inventing itself in real time: specialized tissues, nervous systems, body plans with appendages, eyes, guts. With this explosion of biological novelty came new ecological pressures: predators, pathogens, and symbiotic microbes. Life was no longer a solo act; it became a crowded stage and a simple, hardcoded immune system couldn’t keep up.
To survive in this newly competitive world, organisms needed a flexible immune strategy, one that could recognize unfamiliar threats, adapt to evolving pathogens, and remember past encounters. That was the niche waiting for, even desperate for, adaptive immunity. And the RAG insertion was the catalyst.
How a DNA Hitchhiker Changed Everything
We’ve already described the origin moment: a mobile genetic element—a transposon—inserted itself into the genome of a primitive vertebrate. Most of these genetic stowaways aren’t useful. They barge in, disrupt important genes, and get kicked out or silenced. But this one was different. It brought tools.
It carried genes that encoded enzymes with an unusual skill: they could cut and paste DNA with remarkable precision. Think: molecular scissors with a built-in ruler. This might sound more dangerous than useful, and it could have been, but this time the host survived and evolution took notice.
Instead of evolution wiping out the intruder, organisms with the transposon survived better. Over millions of years, the host genome began to shape it and mutations softened its sharp edges. Regulatory sequences evolved to control where and when the scissors did their cutting. The parasite was slowly domesticated, tamed, and repurposed.
Eventually, it rewired the architecture of immune recognition in vertebrates forever.
Specifically, that insertion enabled a process that lets immune cells shuffle gene segments to create a vast library of unique receptors—each one capable of recognizing a different molecular shape, like a virus or infected cell. These receptors are the key to immune detection, and in the case of B cells, they form the blueprint for antibodies.
Two-Minute Immunology Primer
Let’s take a moment to dust off your pandemic-era immunology knowledge.
Remember antibodies? Those little Y-shaped proteins everyone suddenly had strong opinions about in 2020? They’re not just floating around aimlessly—they’re made by specialized immune cells called B cells, and their job is to recognize, bind to, and help neutralize foreign invaders like viruses and bacteria.
B cells have cousins called T cells, which also play a starring role in your immune defense—responding to infected cells, coordinating responses, and helping make sure the right cells are activated at the right time.
Together, B and T cells belong to a group called lymphocytes, and they have a superpower that makes them unlike any other cells in your body: each one carries a unique molecular sensor—a receptor on its surface that can recognize a specific pathogen or infected cell. In the case of B cells, that sensor does double duty: it helps the cell recognize a threat and, once activated, serves as the blueprint for the antibodies the cell will secrete. These antibodies flood the bloodstream and tissues, each one acting like a lock that’s shaped to catch a specific molecular key—usually a fragment of a virus, bacterium, or other invader.
Their job? To tag invaders for destruction, block their function, or deliver them into the jaws of other immune cells. In short: tag, neutralize, and bag.
Here’s the kicker: your B and T cells can make trillions of different-shaped receptors. Orders of magnitude more than the number of genes you have.
How do they pull this off? That’s where RAG comes in.
Inside every developing B or T cell, long before it ever sees a virus or a vaccine, the RAG proteins go to work. They act like molecular editors, cutting and rearranging small chunks of DNA—modular gene segments called V(D)Js:
V for variable
D for diversity
J for joining
These segments are like genetic Lego bricks. RAG picks one of each, snips them out and glues them together in all sorts of ways, and—voilà—a brand-new, one-of-a-kind receptor blueprint is born. This process is called V(D)J recombination, and it’s the reason your immune system can recognize almost anything, including pathogens it’s never seen before.
This is creativity by design, but also by accident. The DNA gets stitched back together by a kind of cellular duct tape (your DNA repair machinery), which introduces even more variation. Every cell ends up with a slightly different combination.
Most of these receptors won’t bind to anything useful. Some bind to intruders that can harm you. And some might bind to you yourself—to self-molecules. Which brings us to the next critical step in immune development: selection.
Developing B and T cells go through a training program—boot camp in the bone marrow or thymus—where the ones that react strongly to your own tissues are typically destroyed. This process is called central tolerance, and it’s the immune system’s first safeguard against self-attack.
And to think—it all traces back to a molecular accident in the Cambrian Era. A rogue piece of DNA jumped into a genome and, instead of wrecking the place, rewrote the rules of immune recognition. Five hundred million years later, your B cells are still running drills based on that ancient insertion event—shuffling your immune genome and adapting to molecular targets.
So far, so good.
But now comes the question that hangs over all of this: If the immune system can generate trillions of different receptors, how does it avoid turning on its own tissues, to autoimmunity?
The short answer is: it doesn’t. Not always.
The Classical View: How Autoimmunity Slips Through the Guardrails
The very mechanisms that give rise to incredible receptor diversity also open the door to autoimmunity. Some self-reactive cells slip through training. Others mutate later in life. Some stay quiet for years until an environmental trigger wakes them up. Occasionally, the immune system gets confused—mistaking part of your body for an invading pathogen.
This is how researchers believe autoimmune diseases like lupus, rheumatoid arthritis, and autoimmune encephalitis arise.
But it’s not the only possibility. And as we’ll explore later, this entire explanation rests on a framework—a powerful framework that is often predictive, but not a complete one.
Let’s take a moment to introduce the classical model of how autoimmunity arises. This is the theory that underpins nearly every clinical trial, therapeutic strategy, and standard of care today.
It’s a model I know personally. I’ve lived inside its logic. And thanks to therapies based on it, I’m—after adding coffee—a functional human being.
According to the classical model, your immune system is built to distinguish self from non-self. Most self-reactive cells are eliminated during development (this is central tolerance). For those that escape, there’s a backup system—peripheral tolerance—which includes regulatory T cells, immune checkpoints, and protective barriers around sensitive organs like the brain.
Autoimmunity, in this model, arises when:
A self-reactive B or T cell slips through the central and peripheral checkpoints;
It encounters an environmental trigger—like an infection or tissue damage—that activates it; and
It begins to expand and form a clone that mistakenly attacks the body.
This classical model is also known as clonal selection theory, and for over 50 years, it has dominated our understanding of autoimmune disease. It’s largely internally consistent and helps explain why many patients respond to therapies like B cell depletion, T cell suppression, or cytokine blockade.
Like many people with an autoimmune diagnosis, I’ve benefited profoundly from immunosuppressive treatments that emerged from this framework. They’ve pulled me back from the edge more than once.
And yet… the closer you look, the more cracks begin to show in the clonal selection theory of autoimmunity. We have some puzzles that we’ll encounter throughout this series that prompt the following questions:
Why do some healthy people have autoantibodies without any symptoms?
Why do some autoimmune diseases wax and wane?
Why are some diseases so targeted (just one organ) while others affect the whole body?
And why do treatments based on this model sometimes just fail?
These are signs that something deeper is going on—that this classical model, while useful, is incomplete. And that’s where we’re headed in this series. I cannot promise complete answers, but we can journey through the literature together.
Importantly, the classical model didn’t stop us from discovering autoimmune encephalitis, which is the great victory we will be dissecting in the next post. It didn’t stop us from saving lives.
But it has stopped us from connecting what we’ve learned into a broader, unifying theory—one that might not just treat autoimmune disease but transform how we harness the immune system to fight a much wider range of disorders.
In upcoming posts, we’ll examine the cracks in the clonal selection framework, explore ideas like molecular mimicry, degeneracy, pleiotropy, randomness, and the idea that self-recognition isn’t a static boundary, but a dynamic negotiation between immune cells, tissues, and environment.
Stay tuned—and when the day feels heavy, consider this: you are the living legacy of a story that began 500 million years ago with a wandering transposon and a lucky break. It might just add a little bounce to your step.
Good start to this. I'm looking forward to more!
Wow, just wow. I thought that Robert Sapolsky (in the Stanford lectures) was the unrivaled star at explaining biology at the DNA level: he's so clear, moves at exactly the right pace, keeps it interesting, and never says um or ah. You have knocked him off his perch.
The weakest part of these lifesaving and life-changing treatments is that you'd better have a very good medical plan.