

Michael Halassa is professor of Psychiatry and Biomedical Engineering at Virginia Tech. Halassa is also a board-certified and practicing psychiatrist who specializes in treatment of psychotic disorders. His clinical research is focused on identifying novel precision targets based on emerging pharmacology and neurostimulation. He has been consistently funded by the U.S. National Institutes of Health and has received several fellowships and prizes throughout his career—most notably, the Vilcek Prize for Promise in the Biomedical Sciences (2017), an award given to immigrant scientists who have made extraordinary contributions to American society. This essay was originally published on his Substack.
Jaden was twenty-four when I first met him. It was his third admission to an inpatient psychiatric facility due to worsening psychosis. A typical story, in some sense, of medication non-compliance, escalating behavior at home and the family fearing for safety. A typically devastating story too, where just a few years earlier, this mild mannered and pleasant young man was going through high school, with all the promise and hope parents could have for their child. Smart, athletic and with big dreams, ready to take on college and beyond.
Shortly after college had started, Jaden began to struggle. Initially describing that things around him “just didn’t feel right.” He withdrew from his friends, and started missing classes. He then began interpreting ordinary events as carrying hidden meaning directed at him, and stopped leaving his apartment altogether. His roommate found him with a razorblade, cutting into his neck to find the hidden “thought transmitter” that had ruined his life. He did so just in time to call 911 and get him help.
Jaden did not like psychiatric medications. “Dr. Halassa, they just don’t work… I don’t really see the point.” His parents disagreed, and felt that there were some benefits. “You don’t seem to hear the voices anymore, that’s progress.”
“Mom, there is more to this than voices! I need to get my life back!”
And there it was. There is more to life than voices indeed. We do have medications that reduce psychotic symptoms, so from that perspective antipsychotic medications are helpful. However, in many people they do not restore functional capacity. The ability to reason, plan and interact with the world the way they did prior to their first psychotic break. Jaden’s parents agreed: “we would love to have our son back… will we?”
I cannot describe how gut-wrenching these conversations can be. It’s not that there are no examples of people overcoming psychotic illnesses and regaining function. There are, and I did write about that previously and discuss relevant literature. So there is certainly hope for people, but the issue is that we do not yet have a clear way of identifying the pathology in the broad entity we call schizophrenia, nor do we have effective medications that can target the broader syndrome beyond psychosis. To understand just how formidable this task is, I want to walk through what we currently think is going on, where the dominant theory falls short, and what a more complete picture might look like.
Schizophrenia beyond excess dopamine
For most of the past half century, dopamine sat at the center of the pathophysiological models for what we think schizophrenia is. The fact that giving people dopamine augmenting medications (like stimulants) precipitates psychosis and that traditional antipsychotic medications appear to work by blocking dopamine D2 receptors led naturally to the inference that excess dopamine was the problem. Plus, evidence indicated that people early in psychotic illness did have excess dopamine as measured through positron emission tomography (PET).
Shitij Kapur elaborated this connection via the aberrant salience hypothesis, which proposed that dysregulated dopamine signaling causes the brain to assign inappropriate significance to irrelevant stimuli. The world starts to feel charged with meaning that isn’t there. Random events feel connected. The mind builds explanatory narratives to account for these experiences, and because from the patient’s perspective the perceptions are real, those narratives have the internal logic of genuine beliefs.
As many readers of this newsletter know, I don’t particularly love the idea of molecular explanations of mental phenomena, so what are the circuit and computational correlates of this framework? Honestly, I’m not particularly clear on how exactly changes in striatal dopamine are supposed to relate to the experience of psychosis. There are many proposals that relate to the role of dopamine in assigning values to states, but no testable roadmap for how this translates to what people describe as hearing voices. Does this make the connection between dopamine and psychotic symptoms weaker? Possibly, but again, things that tend to increase striatal dopamine tend to increase psychosis (I’ve definitely seen people become psychotic on L-dopa who have never had a history of psychotic experiences, for example) and D2 blockers tend to make these symptoms better.
But the main issue is that schizophrenia is way more than voices, as Jaden pointed out. There are abnormal beliefs which we call delusions, many of which are not particularly ‘fixed’ by D2 blockade. Also, people experience loss of function due to changes in their ability to reason and think clearly. These are the cognitive deficits associated with the disorder which are also associated with ‘negative’ symptoms. Negative symptoms are hard to describe with natural language (you really have to see them to know what they are), but my best attempt at relaying them to you is the following: it’s the lack of initiation of action (and possibly thought), with what appears to be an intact ability to respond when input is provided.
Both cognitive and negative symptoms are linked to functional loss (loss of relationships, employment, education attainment) and both are thought to be a consequence of neural changes that are more causal to the illness than dopamine. Meaning, dopamine dysregulation and the positive symptoms appear to be secondary manifestations of something more fundamental that sits at the core of what we think the schizophrenia(s) are.
The Kraepelinian Core
Emil Kraepelin noticed the central problem more than a century ago, but lacked the tools we currently have to explain it. When he separated dementia praecox from manic-depressive illness, the distinction was primarily prognostic. Manic-depressive patients relapsed and remitted, while the dementia praecox patients deteriorated, often regardless of what happened to their psychosis. Kraepelin’s insight was that the trajectory was the illness, not the psychotic episodes that waxed and waned. He thought that an illness core may be eroding function steadily, in a way that was independent of psychosis altogether. That observation has held up across more than a century of follow-up data, and it is part of what makes schizophrenia so different from many other conditions we treat in psychiatry.
The evidence accumulated since Kraepelin points, imperfectly but consistently, toward the prefrontal cortex and the circuits that support how it does its work. The prefrontal cortex is responsible for the kinds of thinking that is most prized by us humans: maintaining goals across time, adapting to changing contexts, filtering out irrelevant information, and making decisions under uncertainty. Like all other cortical areas, the prefrontal cortex is built from both excitatory and inhibitory neurons; excitatory neurons are the main conduits of signals as they can drive one another (to sustain working memory or integrate signals across time for example) and project instructions to other areas, whereas inhibitory neurons control the activity patterns locally. There are many more excitatory neurons than inhibitory ones, but inhibitory function is critical for maintaining appropriate operating regime in cortical circuits, including operations like signal amplification and noise suppression. People have coined the term Excitatory/Inhibitory Balance (which was really developed for synaptic inputs on individual neurons, but gets used a lot more liberally to describe macroprocesses too) as a set point parameter for circuit function. In schizophrenia, converging evidence from postmortem studies, neuroimaging, and animal models points toward disruptions in inhibitory control that may give rise to loss of signal fidelity and accumulation of neural noise.
This formulation makes sense, broadly, when thinking about reasoning in schizophrenia and why it may be difficult for people to think clearly. Our lab has done a bit of work in this area (but we’ve also followed the literature very closely so it’s not just one experiment I’m highlighting). What we have observed is that patients with schizophrenia perform comparably to healthy individuals when task demands are straightforward, but some fail disproportionately as ambiguity increases. One view is that this is the consequence of a perturbed prefrontal network with particular failure modes that makes the brain susceptible to a variety of changes in the ability to reason, plan and initiate action. It also changes the ability to control downstream circuits including the dopaminergic system. Again, classical antipsychotic medications are able to address the dopaminergic abnormalities but do very little in addressing the prefrontal changes that may be at the core of these illnesses.
Schizophrenia may be hard to treat because we are not getting to its core.
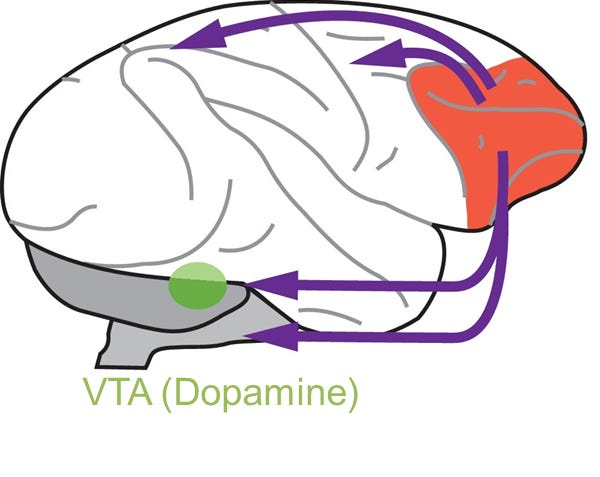
Image illustrating prefrontal ‘top down control’ (prefrontal cortex: red), including that of dopaminergic function (ventral tegmental area: green). Modified from Ott and Nieder, 2019 TICS
Psychosis is not schizophrenia
If dopamine dysregulation is downstream of a more foundational core of schizophrenia, shouldn’t we be re-thinking the relationship between the two? Rather than being the defining feature of the illness, it becomes a clue: evidence that something has failed to regulate the dopaminergic system, resulting in surges that are associated with psychotic experiences. And one of the more interesting things about those surges is that they can apparently be triggered by multiple pathways.
Psychotic mania, which can happen in bipolar disorder, is a useful example. Molecular imaging studies find elevated dopamine receptor availability specifically in psychotic mania, with no significant difference between non-psychotic mania patients and healthy controls. A PET imaging study comparing bipolar disorder with psychosis directly to schizophrenia found that the dopaminergic signature in bipolar psychosis was comparable to what is seen in schizophrenia.
ADHD offers yet another route. In a subset of patients, particularly with high-dose amphetamines, psychosis can emerge. What’s intriguing about this is that there is a high co-occurrence of ADHD and bipolar disorder, which appears to be innate rather than treatment-driven. This suggests that some patients may carry a genetic liability spanning both conditions, placing them closer to the threshold to begin with. The diagnostic complexity these patients present clinically may reflect something important about their underlying biology that we have yet to understand.
What all of this suggests is that psychosis is better understood as a state than as the defining feature of any particular illness. In schizophrenia, the push toward that state may come from disrupted prefrontal function. In bipolar disorder and in stimulant-induced psychosis, it may come from other underlying mechanisms. This is a very important set of research questions that we need both public investment in and new talent to continue the quest of answering them.
But can excess dopamine drive schizophrenia?
This brings me to a question I find quite unsettling, and one the field has not fully worked through. If schizophrenia involves primary circuit disruption that produces dopamine dysregulation as a downstream consequence, what happens when you invert the sequence? When you start with dopamine and drive it hard enough, for long enough, we may be arriving at something that begins to resemble the underlying pathology we see in schizophrenia.
The animal literature on chronic amphetamine exposure offers some evidence that you can, at least partially. When dopamine is repeatedly and forcefully elevated, a secondary deficit in prefrontal function appears to emerge as a consequence. The cognitive deficits that follow, particularly in the kinds of flexible contextual thinking that depend on intact prefrontal function, begin to resemble what we observe in schizophrenia. In humans, chronic heavy methamphetamine use can produce persistent syndromes that extend well beyond the period of active drug use and include not just psychosis but negative symptoms and cognitive impairment. Strikingly, these cases are indistinguishable from schizophrenia that emerges the way it did in Jaden.
What it suggests is that dopamine dysregulation is not only a consequence of the primary pathology. When it is severe and sustained enough, it may begin to produce something like that pathology from the other direction. Whether that circularity has implications for how we think about illness progression in patients like Jaden, who experience repeated psychotic episodes with incomplete recovery between them, is very important. Because it helps inform whether psychotic episodes have a kindling effect, or not. Meaning, if people are slipping into psychosis (because of medication non-compliance for example), and when they are out of it, their frontal function is worse off than before, they need to know that so they are clear on the cost-benefits of taking medications, no matter how imperfect they currently are.
Map showing global prevalence of methamphetamine use. From this Lancet article.
For schizophrenia prevalence, check out this interactive map here.
Reasons for optimism
Jaden’s frustration was spot on. The voices were the part of his illness that our treatments could reach. They were not the part that determined whether he would get his life back.
Schizophrenia may be hard to tackle because psychosis, the feature that defines it in most people’s minds and that drives most of its treatment, is not the most devastating part of the illness. The illness may live in circuits that support how a person thinks, adapts, and engages with the world, circuits that were already showing signs of disruption before the first psychotic break and that tend to remain disrupted after psychotic surges are brought under control. The gap between what antipsychotics can do and what patients and families are actually asking for may be a consequence of having organized the entire treatment framework around a downstream signal while the upstream pathology has largely gone unaddressed.
But there is lots to be optimistic about.
The most significant development in schizophrenia pharmacology in decades is KarXT (Cobenfy), approved by the FDA in September 2024. Cobenfy combines xanomeline, a muscarinic cholinergic receptor agonist, with trospium chloride, which prevents xanomeline from acting outside the central nervous system and causing intolerable side effects. What makes it historically notable is simple: it is the first drug approved for schizophrenia in over seventy years that does not work by blocking dopamine D2 receptors. Cobenfy targets muscarinic M1 and M4 receptors, thought to act upstream of dopamine. M4 which is distributed subcortically, impacts dopamine release (and psychotic symptoms), while M1 which is enriched in the prefrontal cortex may get closer to the Kraepelinian Core of the illness.
This is consistent with both placebo controlled clinical trial data showing improvements on both negative and cognitive symptoms as well as preliminary real world reports. While we still need larger trials to confirm, the idea is that Cobenfy may be doing something different from traditional antipsychotics and that the patients most likely to benefit from that difference can be identified clinically. It is certainly going at least one step upstream of D2 blockers.
Other pharmaceutical companies are trying even further upstream, to target mechanisms of prefrontal excitatory/inhibitory balance more directly. One example I am familiar with is Ovid Therapeutics, which is developing a class of compounds that directly activate KCC2, the potassium-chloride cotransporter that controls chloride concentrations inside neurons. Chloride homeostasis is what determines whether GABA, the brain’s primary inhibitory neurotransmitter, can work effectively. Postmortem studies in schizophrenia have found reduced KCC2 in the prefrontal cortex, and animal models of schizophrenia-like pathology (in our hands) replicate the finding. Restoring KCC2 function would, in principle, address the inhibitory deficit directly rather than compensating for its downstream consequences. The first intravenous KCC2 activator, OV350, completed Phase 1 safety testing in late 2025 with a clean safety profile and electrophysiological signals consistent with central activity. An oral candidate, OV4071, is entering clinical trials in 2026, and will be applied to schizophrenia in 2027 if all goes well. I recently participated in an Ovid event and talked about our work here (you just need to enter some info to gain access to the full recording). Oliver (Ollie) Howes also spoke right before me and introduced the prefrontal E/I balance framework in relationship to his work.
None of this means we are close to having something to offer Jaden that addresses what he was asking for. The gap between what these drugs might eventually do and what he needed in that room remains large. But for the first time in a long while, the therapeutic programs being developed are starting to engage the biology that the evidence points toward, rather than cycling through variations on the same dopamine-blocking approach that has dominated since the 1950s. That is the right direction and for now, it’s the best we are all able to do.
If you found this piece useful, please consider subscribing and sharing it with someone who might find it interesting. Paid subscriptions help support the time it takes to produce this work.
Disclaimer: Jaden is a fictional character who draws upon a composite of patients I’ve interacted with over the years. I cannot overstate the sense of privilege I feel to have been a part of these folks’ and their families’ lives.
Great read and somehow hopeful!
Excellent read, thank you! Regarding the correlations observed between ADHD and bipolar you mention specifically, I thought there had been robust enough genetic findings regarding COMT gene expression, and its most well-known mutations (fast and low COMT vs. "normal" COMT), to support this? The associated phenotypes clearly underpin the aforementioned correlations (as well as any you mention that involve dopamine dysregulation, potentially). Again, not the whole picture, but an important piece perhaps?