
From Clinic to Genome and Back Again: Cowden Syndrome and PTEN
Author: Stetson Thacker

Stetson Thacker completed a PhD at Case Western Reserve University in genomic medicine. He currently works in industry supporting and developing tools for advancing precision oncology care and research. He has a broad range of interests, ranging across the sciences and humanities, that he enjoys opining on.
This is the first post in a series on hereditary cancer/cancer syndromes on Stetson’s blog Holodoxa.
In 1963, two young internal medicine residents, Kenneth M. Lloyd II and Macey Dennis, from Youngstown, Ohio reported a striking case study of a 20-year-old female with “a new familial disease” of “multiple congenital defects,” who was brought in for evaluation because of a persistent lesion on her right breast. In an era before HIPAA, Lloyd and Dennis opted to name this new syndrome after the patient herself, Rachel Cowden. Little did they know, at the center of their clinical discovery hid one of the most important cancer-related genes in the human genome, PTEN.
Rachel and the First Cowden Family
Lloyd and Dennis’ case study of Rachel Cowden presents a thorough examination of her “unusual symptom complex.” This catalogue of Cowden traits is assembled in medical jargon so I’ll translate and reproduce some images from their report.

Rachel was described as having “adenoid facies,” which referred to the long, narrow, and open-mouthed appearance of her face. This was accompanied by other related features like an underdeveloped jaw and oral cavity. Lloyd and Dennis also described her as “asthenic-appearing,” referring to her slender build. She had a concave chest and curved spine, musculoskeletal abnormalities often associated with genetic disease. Rachel was also found to have growths on her lips, tongue, thyroid, bones, and breasts. Her breasts were enlarged and discolored, though she had never been pregnant. She had previously been diagnosed with fibrocystic disease of the breast, meaning the fibrous tissue there was growing abnormally, creating a cobblestone texture. There were early signs of cancer in her breast tissue too. Rachel was also reported to have neurodevelopmental issues, including intellectual deficits (an IQ of 85) and poor motor coordination.

Notably, Rachel’s family members shared similar traits, though not quite as severe as her. Hence, Lloyd and Dennis described the family history as “forme fruste,” meaning attenuated in presentation. In hindsight, the clinical phenotypes (i.e. traits) recorded are a big flashing sign that this is genetic disease. Something that did not escape the notice of Lloyd and Dennis.
Rachel’s mother (55) had a large goiter and “a marked emotional stutter.”
Rachel’s sister, deceased due to an accident, had thyroid and skin tumors and intellectual deficit.
Rachel’s living sister (24) appeared normal.
Two of her three maternal aunts died of breast cancer and the surviving aunt was institutionalized due to a diagnosis of “post-encephalitic Parkinsonism” and reported to have similar facial and neurological phenotypes to Rachel.
Five maternal uncles and father appeared unaffected by similar symptoms.
Analysis of her chromosomes found no abnormalities though Lloyd and Dennis remained confident the cause was genetic. The DNA origins of Rachel’s symptoms would have to await the maturation of clinical genetics (the structure of DNA was only first described a decade earlier). After her evaluation, Rachel was treated with a bilateral mastectomy (removal of both breasts) and a thyroidectomy (removal of the thyroid gland). Unfortunately, she died of metastatic breast cancer roughly a decade later.
The Tumor Suppressor Gene on Chromosome Ten, PTEN
For roughly three decades after the first description of Cowden Syndrome, the responsible genetic lesion remained elusive. Nonetheless, similar clinical cases continued to be documented with an expanding range of phenotypes, including macrocephaly (an abnormally large head), benign growths called “hamartomas,” and cancers affecting tissues beyond the breast and thyroid. Finally, the case was largely resolved by two seminal papers published in Nature Genetics in 1996 and 1997. Together, these studies identified a gene called PTEN or the Phosphatase and TENsin homolog on chromosome 10 as the culprit behind Cowden Syndrome (or at least a substantial portion of cases, though we’ll get to this later).
PTEN Uncovered in Cowden Syndrome
A 1996 study titled “Localization of the gene for Cowden disease to chromosome 10q22-23” was the product of a fruitful international collaboration among European labs steered by the leadership of Dr. Charis Eng. It used what’s called genetic linkage analysis to identify a chromosomal region that associated strongly with Cowden syndrome diagnosis by studying 40 Cowden patients from 12 families.
Genetic linkage analysis cleverly leverages the process of genetic recombination, the exchange of different DNA segments between matching, nonidentical chromosomes, maternal and paternal pairs. Recombination occurs during sex cell formation, a process called meiosis. Linkage studies follow recombination events within families affected by a disease or a given trait in order to generate an odds score that conveys the likelihood that some segment of DNA is linked to the disease/trait of interest. In other words, linkage analysis is just the tracking of signposts throughout the genome in related people to see if certain signposts frequently appear in association with the phenotype of interest or not. Once complete, geneticists can confidently point to a modestly sized chunk of DNA and say it’s important to a trait/disease process. In the linkage study led by Dr. Eng, the team found that a part of the long arm of chromosome 10 is many many million times more likely than not to be linked to Cowden Syndrome! However, there were several candidate genes in this region that could have been relevant to Cowden Syndrome so a more detailed map was needed.

The follow-up 1997 study, also led by Dr. Eng, more finely mapped the linked region to “a narrow interval less than 1 cM” or a span of 1 million base pairs of DNA. A million base pairs may sounds big, but, for reference, the average gene spans 24 thousand base pairs, and only ~25% of the genome is made of gene sequences. This region was home to a gene called PTEN. And PTEN made for a great candidate gene for a genetic cancer syndrome because just months earlier another study in Nature Genetics identified PTEN as tumor suppressor gene in sporadic cases of cancer. A tumor suppressor gene is a gene that is shown to be important to keeping normal cells from transforming into cancer. There is a common metaphor used by those teaching about cell growth and division of gas pedals vs brake pedals, where (you guessed it) tumor suppressors are the brakes and genes called oncogenes are the gas pedals. Not so surprisingly, the alternative name for PTEN used by the aforementioned study in sporadic cancer was MMAC1, which stood for “Mutated in Multiple Advanced Cancers 1.”
The 1997 study did more than just more than narrow down the region, the team of scientist performed an analysis of the PTEN sequence itself, assessing the individual base pairs in affected patients compared to unaffected controls. They found several changes in the DNA, the As, Cs, Gs, and Ts of PTEN, in Cowden patients. Geneticists call these changes mutations, variants that deleteriously alter a gene’s function. Specifically, three different mutations were observed in four of the five Cowden families (see Table).
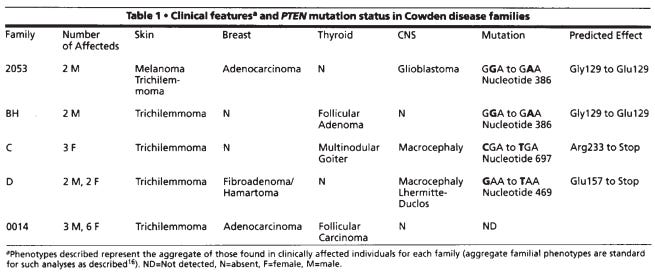
Two were changes that were expected to completely destroy the function of PTEN, Glu157 to Stop and Arg233 to Stop. These types of mutations are called nonsense variants or null alleles because no protein product is expected to result; thus, the gene can not have its normal effect. The other two mutations were the exact same substitution at the 386th nucleotide from a G to an A in PTEN’s coding sequence, the part of the gene that gets turned into protein. This change converted the amino acid glycine at position 129 in the PTEN protein to glutamine. This change was expected to be damaging because it affected a part of the protein that performs an important chemical reaction (removes a phosphate group from proteins and lipids) and is shared by other proteins that perform similar functions. The authors phrase this as, “the codon Gl29E missense mutation observed in families 2053 and BH occurred within the signature sequence found in all protein tyrosine phosphatases and dual-specificity phosphatases.”
Several other studies replicated these findings. Altogether this was persuasive evidence that PTEN is a definitive risk gene for Cowden Syndrome. These foundational findings have inspired an additional two-and-a-half decades of research on PTEN and related biology, affecting the care and management of both patients with Cowden Syndrome and those with sporadic cancers that harbor PTEN mutations.
The Problem of Clinical and Genetic Heterogeneity
The discovery of PTEN and its connection to cancer was important, but it didn’t answer all the pressing questions about Cowden Syndrome and a cropping of other syndromes with similar complexes of symptoms.
As may have been apparent from the above, not all Cowden patients carry a PTEN mutation. There are now many reports that Cowden Syndrome can result from (or at least is associated with) mutations in other genes (PIK3CA, AKT1, SEC23B) and hypermethylation of the promoter of a gene adjacent to PTEN, KLLN. In some ways this isn’t surprising, Cowden as a clinical entity is a bit unwieldy. It has a litany of associated symptoms and complex diagnostic criteria. It’s enough to flummox many competent physicians. Subsequently, Cowden Syndrome is likely under-diagnosed.
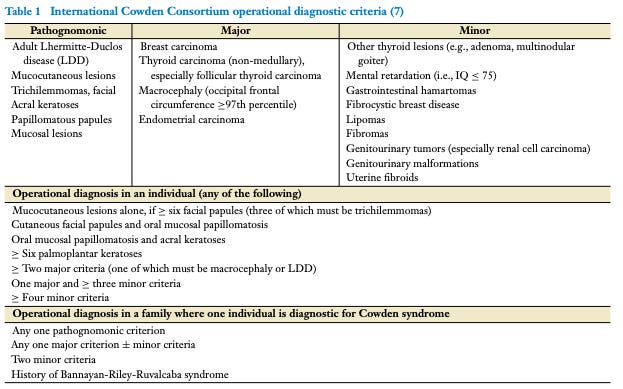
Also previously mentioned, there are a number of other clinical diagnoses, other cancer or neuro-developmenal syndromes, that overlap with Cowden Syndrome: Bannayan-Riley-Ruvalcaba syndrome (BRRS), Proteus syndrome (PS), Proteus-like syndrome (PLS), Juvenile polyposis syndrome (JPS), and Peutz-Jeghers syndrome, Neurofibromatosis type 1, etc. Plus, inherited mutations in PTEN are often observed in some of these syndromes. To resolve this heterogeneity, a molecular diagnosis was forwarded by PTEN-whisperer Dr. Eng: PTEN Hamartoma Tumor Syndrome or PHTS. PHTS encompasses CS, BRRS, PS, and PLS by including any symptomatic individual who is found to have a PTEN mutation in their germline.1

The value of a consolidating molecular diagnosis, the PTEN mutation, is that it enabled a more robust understanding of the clinical spectrum of PHTS. Relatedly, it enabled study of genotype-phenotype relationships, the connections between the genetic information (genotype) of an organism and its physical traits (phenotype). For a PHTS patient, there are a number of potential health issues that may present over his or her lifetime and clinicians needs to know the general risk for each and beyond that have some understanding of what to expect for a specific patient given his or her specific PTEN mutation, i.e. deliver real personalized medical care. The reason that this is thought to be possible is that PHTS patients can be roughly classed into two groups: those with problems of neurodevelopment like autism and those with cancer and benign overgrowth. There are of course patients affected by both sets of phenotypes (like Rachel), but this variability suggests it’s possible that different types of PTEN mutations can steer these outcomes. I’s also likely other genes and variables2 may shape these outcomes too. This approach has provided a great deal of clinical success. These insights are now codified in professional practice guidelines (see Table).

A Triumph of Translation: Precision Genomic Medicine
Our current clinical management of PHTS exemplifies the power of genetic science. Scientists used the methods of genetics (e.g. linkage analysis and DNA sequencing) to identify the cause of a cancer syndrome with a complex spectrum of phenotypes. The genetic diagnosis helped parse the clinical complexity and enable a precision approach to the care of PTEN patients.
Stay tuned for the next article in the series where I dive deeper into what PTEN actually does and how it is relevant to cancer processes.
The germline line is essentially the inherited genome. This includes a little more variation than just the respective genomic contributions from each parent as new or de novo changes occur in the parent “germ” or sex cells or during embryonic development. And there have been several cases of de novo PTEN mutations in Cowden and related syndromes.
As you may have noticed, I’ve elided the issues of sex a bit. Male PHTS cases tend to have autism and other neurodevelopmental issues more frequently than female PHTS cases, while cancer risk is higher in the female PHTS cases. However, it isn’t entirely known exactly what explains this, and if this observation completely forecloses the impact of different PTEN mutations on the PHTS clinical spectrum.